
Đang tải câu hỏi…
Quiz Hoàn Tất!
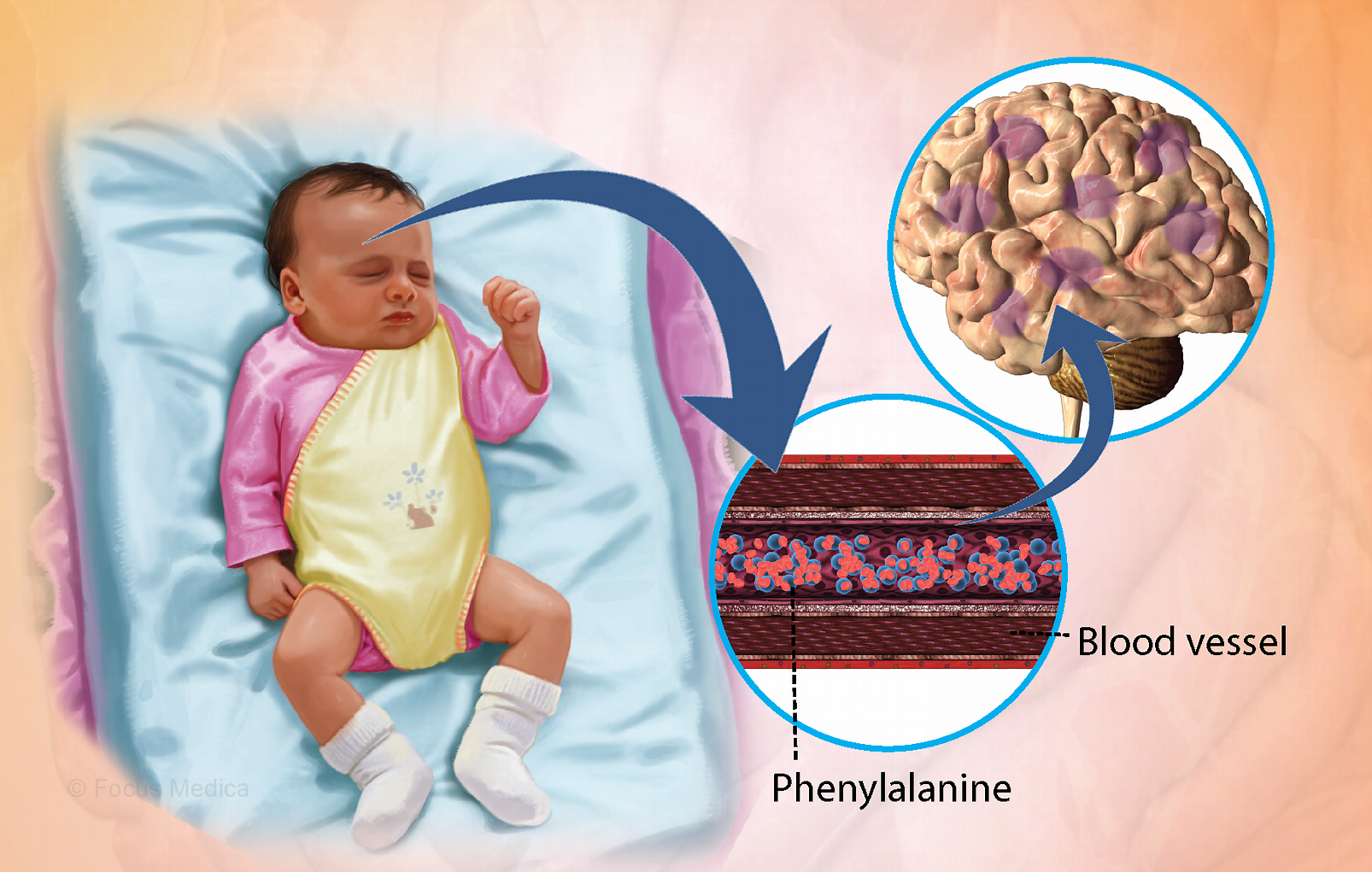
1. Bệnh Phenylxeton niệu (Phenylketonuria – PKU)
Đây là bệnh rối loạn chuyển hóa axit amin Phenylalanine, phổ biến ở người da trắng (Tần suất 1/10.000).
Nguyên nhân và Cơ chế
- Di truyền: Do gen lặn nằm trên nhánh dài NST thường số 12.
- Cơ chế: Cơ thể thiếu hụt enzyme PAH (Phenylalanine hydroxylase).
- Bình thường: PAH giúp chuyển hóa Phenylalanine
→Tyrosine. - Khi bệnh: Phenylalanine không chuyển hóa được sẽ ứ đọng trong máu và mô
→\rightarrow→đầu độc hệ thần kinh trung ương. - Thiếu Tyrosine dẫn đến thiếu Melanin (sắc tố màu).
- Bình thường: PAH giúp chuyển hóa Phenylalanine
Triệu chứng nhận biết
- Ngoại hình: Da trắng, tóc vàng, mắt xanh (do thiếu melanin).
- Thần kinh: Trẻ kích động, co giật, tăng phản xạ.
- Trí tuệ: Chậm phát triển ngôn ngữ và trí tuệ nghiêm trọng (IQ < 20), đầu nhỏ.
- Nước tiểu: Có mùi mốc (do bài xuất phenylpyruvic acid).
Chẩn đoán và Điều trị
- Sàng lọc: Xét nghiệm mức phenylalanine trong máu (lấy máu gót chân 48-72h sau sinh, khi trẻ đã ăn sữa).
- Điều trị: Ăn kiêng thực phẩm chứa Phenylalanine suốt đời và bổ sung Tyrosine.
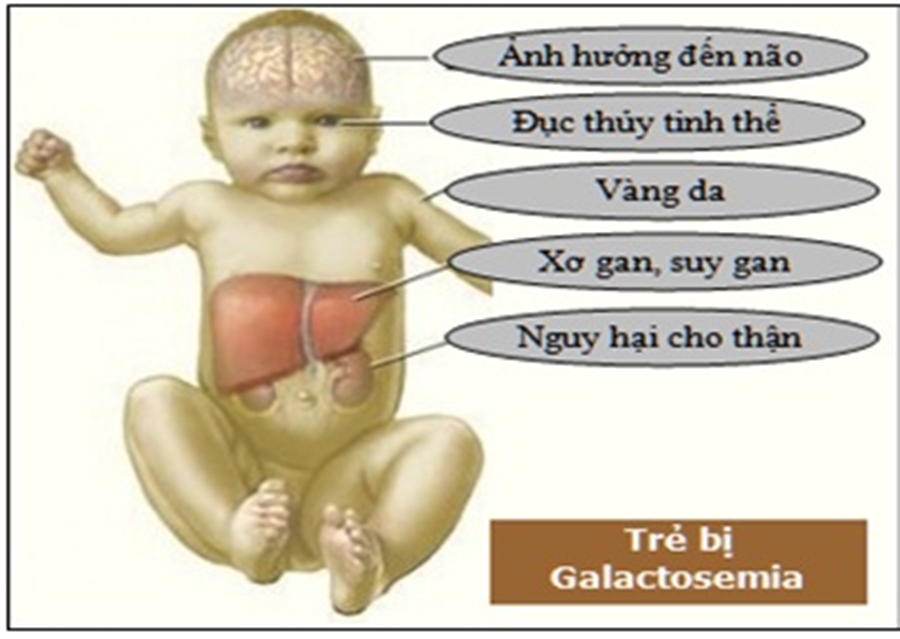
2. Bệnh Galactose huyết (Galactosemia)
Đây là bệnh rối loạn chuyển hóa Carbohydrate (đường), khiến cơ thể không tiêu thụ được đường Galactose (có nhiều trong sữa).
Các thể bệnh và Nguyên nhân
Quy luật di truyền: Alen lặn trên NST thường. Có 3 thể bệnh chính do thiếu hụt 3 loại enzyme khác nhau:
- Thể cổ điển (Nặng nhất): Do thiếu enzyme G1PUT (gen trên NST số 9). Gây tích tụ độc tố làm tổn thương gan, não, mắt.
- Do thiếu Galactokinase: Gen trên NST số 17.
- Do thiếu UDP-epimerase: Rất hiếm gặp.
Triệu chứng lâm sàng
Trẻ sinh ra bình thường nhưng sau khi bú sữa vài ngày/tuần sẽ xuất hiện:
- Nôn mửa, không tiêu được sữa, suy dinh dưỡng.
- Đục nhân mắt (Cataract): Triệu chứng điển hình do tích tụ galactiol trong thủy tinh thể.
- Xơ gan, chậm phát triển trí tuệ.
- Ở nữ giới: Buồng trứng không phát triển.
Điều trị
- Chế độ ăn kiêng tuyệt đối: Loại bỏ hoàn toàn Galactose (không dùng sữa mẹ và các loại sữa chứa lactose/galactose).
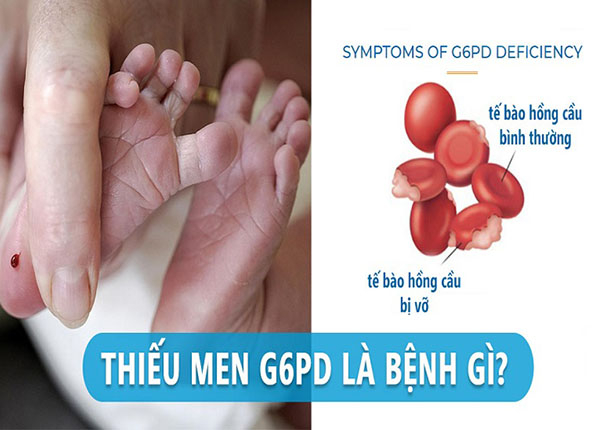
3. Bệnh thiếu men G6PD (Glucose-6-phosphate dehydrogenase)
Đây là bệnh di truyền phổ biến nhất, với khoảng 400 triệu người mắc trên thế giới (Châu Á chiếm khoảng 2%).
Vai trò của G6PD và Cơ chế bệnh
- Vai trò: G6PD là enzyme quan trọng giúp màng tế bào hồng cầu bền vững trước các tác nhân oxy hóa.
- Di truyền: Gen lặn nằm trên NST giới tính X (Xq28).
→Nam giới dễ mắc bệnh hơn nữ giới. - Cơ chế: Khi thiếu men G6PD, hồng cầu mất khả năng bảo vệ. Khi gặp các chất oxy hóa mạnh (thuốc, thức ăn...), hồng cầu sẽ bị vỡ (tan máu).
Triệu chứng
Bình thường người bệnh không có biểu hiện. Cơn tan máu cấp tính chỉ xuất hiện sau 2-3 ngày dùng các tác nhân kích thích:
- Sốt, đau đầu, đau bụng, đau thắt lưng.
- Vàng da, niêm mạc nhợt nhạt.
- Nước tiểu màu nâu đen (tiêu biểu của tan máu).
- Nặng có thể dẫn đến hôn mê, suy thận.
Các tác nhân cần tránh
Người bệnh cần tuyệt đối tránh các loại thuốc có tính oxy hóa:
- Thuốc sốt rét (Quinine, Primaquine...).
- Kháng sinh nhóm Sulfonamide, Nitrofurantoin.
- Các loại thuốc giảm đau hạ sốt (Aspirin, Pyramidon...).
- Tránh ăn đậu tằm (Fava beans) và tiếp xúc với băng phiến (long não).
Phòng bệnh
- Sàng lọc sơ sinh để phát hiện sớm.
- Người mang gen bệnh cần luôn giữ bên mình danh sách thuốc cấm sử dụng.
Tóm tắt so sánh nhanh
| Đặc điểm | PKU (Phenylxeton niệu) | Galactosemia (Galactose huyết) | Thiếu men G6PD |
| Rối loạn chất | Axit amin (Protein) | Đường (Carbohydrate) | Enzyme bảo vệ hồng cầu |
| Di truyền | Lặn - NST thường (12) | Lặn - NST thường (9 hoặc 17) | Lặn - NST giới tính X |
| Hậu quả chính | Ngốc nghếch (IQ thấp), da trắng bệch | Đục nhân mắt, xơ gan | Tan máu (vỡ hồng cầu) khi uống thuốc lạ |
| Điều trị/Phòng | Ăn kiêng đạm (Phenylalanine) | Kiêng sữa (Galactose) | Tránh thuốc oxy hóa |
Comment ×