Để hiểu về bệnh, ta cần biết về Hemoglobin (Hb) - huyết sắc tố giúp hồng cầu vận chuyển oxy:
Hb bình thường gồm 4 chuỗi globin kết hợp với nhân Heme.
Ở người trưởng thành ( HbA1HbA_1HbA1 ), cấu trúc gồm: 2 chuỗi α (alpha) + 2 chuỗi β (beta).
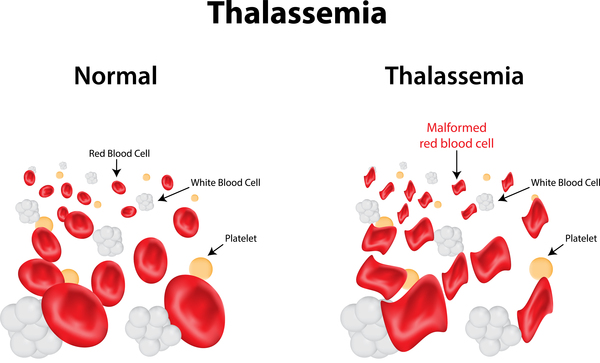
Nguyên lý gây bệnh: Khi gen bị đột biến làm giảm hoặc mất hẳn khả năng sản xuất một loại chuỗi (Alpha hoặc Beta), sự cân bằng bị phá vỡ, hồng cầu dễ bị vỡ (tan máu) → Thiếu máu mãn tính.
2. Phân loại bệnh Thalassemia
Dựa vào loại chuỗi globin bị thiếu hụt, bệnh được chia làm 2 nhóm chính:
2.1. Alpha-Thalassemia ( α -Thalassemia)
Đây là tình trạng thiếu hụt chuỗi α-globin.
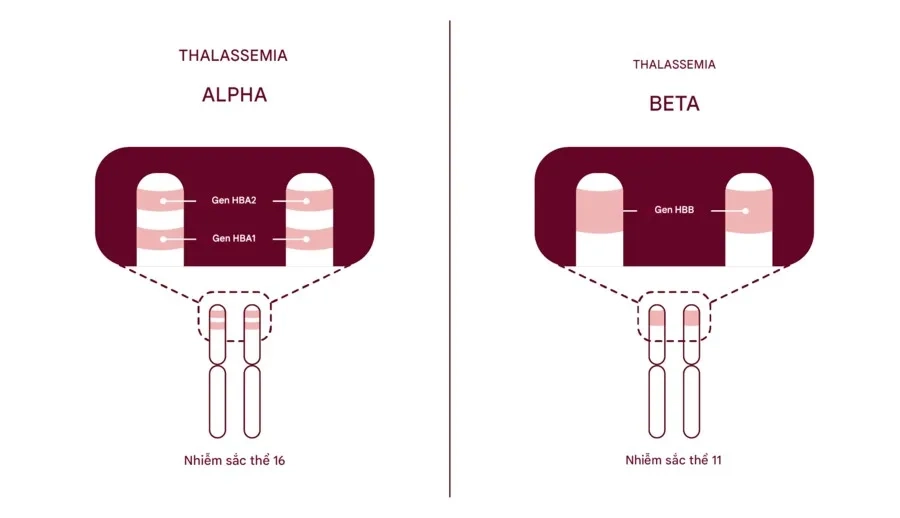
Vị trí gen: Gen quy định chuỗi α\alphaα nằm trên nhánh ngắn của NST thường số 16.
Số lượng gen: Mỗi người bình thường có 4 gen α (2 gen trên mỗi NST 16).
Cơ chế đột biến: Chủ yếu là do MẤT ĐOẠN GEN.
Các mức độ bệnh (Tương ứng số gen bị mất):
Mất 1 gen: Người lành mang gen bệnh, không biểu hiện triệu chứng.
Mất 2 gen: Thiếu máu nhẹ, hồng cầu nhỏ.
Mất 3 gen (Bệnh HbH): Thiếu máu vừa hoặc nặng. Cơ thể tạo ra HbH (gồm 4 chuỗi β ).
Mất 4 gen (Hb Bart’s): Thể nặng nhất. Cơ thể không tạo được chút chuỗi α nào. Thai nhi bị phù nề, thường tử vong ngay trong bụng mẹ hoặc ngay sau sinh.
2.2. Beta-Thalassemia ( β -Thalassemia)
Đây là tình trạng thiếu hụt chuỗi β-globin.
Vị trí gen: Gen quy định chuỗi β nằm trên nhánh ngắn của NST thường số 11.
Số lượng gen: Mỗi người có 2 gen β (1 gen trên mỗi NST 11).
Cơ chế đột biến: Chủ yếu là ĐỘT BIẾN ĐIỂM (thay thế, thêm hoặc mất nucleotide) dẫn đến:
β+ : Giảm khả năng tổng hợp.
β0 : Mất hoàn toàn khả năng tổng hợp.
Cơ chế sinh bệnh đặc trưng: Khi thiếu chuỗi β , các chuỗi αdư thừa sẽ kết tụ lại → làm vỡ hồng cầu → Thiếu máu. Để bù đắp, tủy xương phải hoạt động quá mức gây biến dạng xương.
Các thể lâm sàng:
Thể nhẹ (Trait): Mang 1 gen bệnh, không triệu chứng.
Thể trung gian: Thiếu máu mức độ trung bình.
Thể nặng (Bệnh Cooley): Biểu hiện rất sớm và nặng nề.
3. Triệu chứng nhận biết và Hậu quả
Đặc biệt ở thể nặng (Beta-Thalassemia thể Cooley), bệnh nhân sẽ có các biểu hiện điển hình:
Biến dạng xương (Vẻ mặt Thalassemia): Do tủy xương tăng sinh mạnh để tạo máu, làm xương sọ to, trán dồ, gò má cao, mũi tẹt.
Gan lách to: Do phải làm việc quá tải để lọc hồng cầu vỡ và tạo máu ngoại vi.
Ứ sắt: Do truyền máu nhiều lần và tăng hấp thu sắt, dẫn đến sạm da, suy gan, suy tim, suy tuyến nội tiết.
4. Điều trị và Phòng ngừa
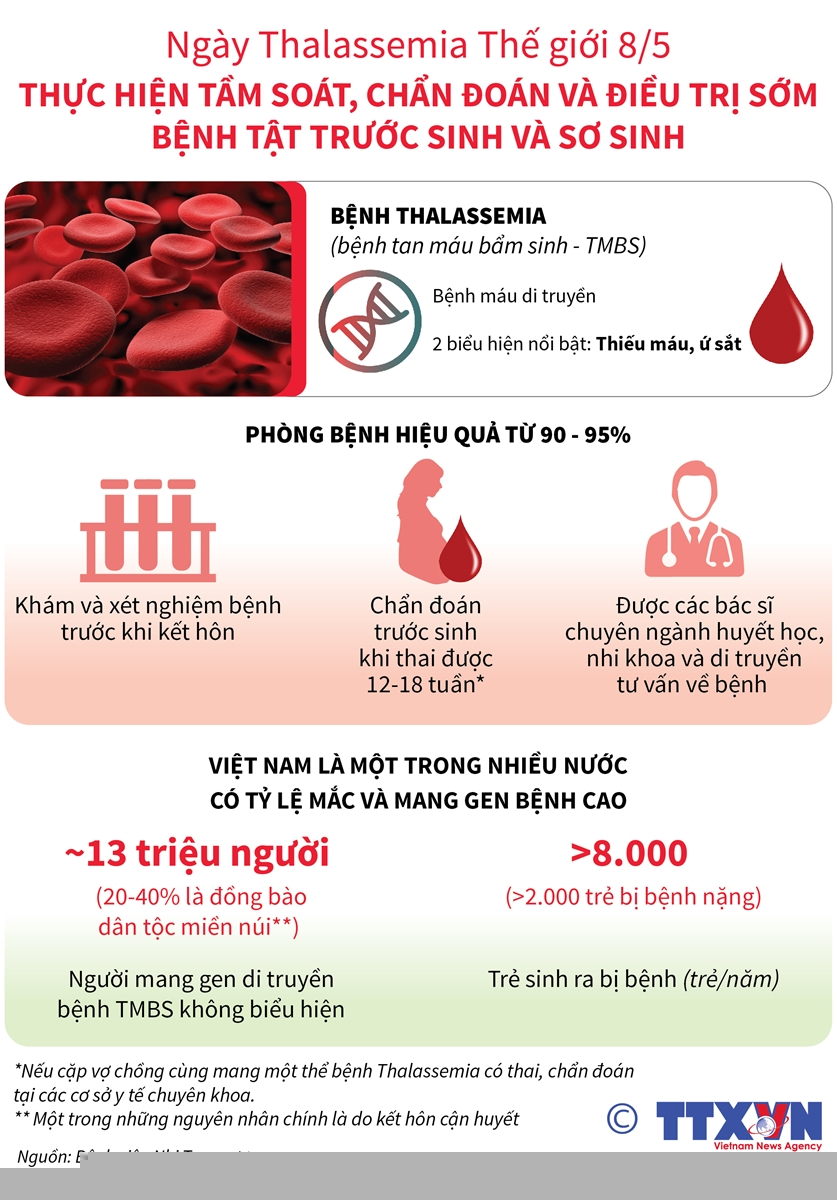
Thalassemia là bệnh di truyền lặn trên NST thường, nghĩa là nếu cả Bố và Mẹ đều mang gen bệnh (dù là người khỏe mạnh), con cái sinh ra có 25% nguy cơ mắc thể nặng.
Điều trị:
Truyền máu định kỳ suốt đời (đối với thể nặng).
Thải sắt (dùng thuốc để loại bỏ sắt thừa khỏi cơ thể).
Ghép tủy xương (phương pháp duy nhất có thể chữa khỏi nhưng chi phí cao và khó tìm người cho phù hợp).
Phòng bệnh (Quan trọng nhất):
Tầm soát tiền hôn nhân: Xét nghiệm công thức máu và gen để biết mình có mang gen bệnh hay không.
Chẩn đoán trước sinh: Nếu bố mẹ cùng mang gen, cần chẩn đoán sớm cho thai nhi.


Comment ×